芙蓉姐翻译
非小细胞肺癌(NSCLC)患者在接受nintedanib(N)和多西他赛(D)治疗中的抗血管生成特定的不良事件(AEs)(2014 美国临床肿瘤学会)
摘要
背景:抗血管新生治疗,包括单克隆抗体和TKIs,显示肿瘤的活动; 然而,他们有限的使用部分是由于特性的副作用(如出血、血栓形成、穿孔、严重皮肤反应、高血压)。N (nintedanib)口服,每日两次,是三重angiokinase抑制剂。我们扩展我们的调查LUME-Lung 1研究(临床试验••gov NCT00805194)和评估添加N标准二线D是否增加抗血管生成药物关联的特性副作用发生的频率和这些副作用是否限制了使用N . 方法 UME-Lung 1,一项随机、安慰剂对照III期试验研究N + D在晚期非小细胞肺癌患者的一线化疗失败后,显著地提高PFS不论组织学,显示明显提高患者的腺癌生存。抗血管生成关联的 AEs的发病率和强度根据常见术语标准AEs(CTCAE版本3.0)评估所有病人至少接受一剂N,D,或安慰剂(Pl)。结果VEGF抑制剂AEs更常见(≥2%的差异)在N和Pl组出血(所有等级:14.1% vs 11.6%;等级≥3:2.3% vs 1.8%)和高血压(所有等级:3.5% vs 0.9%;等级≥3:0.6% vs 0.2%)。当我们比较组织学抗血管生成关联 AEs的差异时,我们发现了名义上的差异。更多流血事件被报道为N -治疗鳞状细胞癌(SCC)患者(所有等级:17.1% vs 10.9%;等级≥3:2.9% vs 1.3%)超过那些腺癌(所有等级:10.9% vs 11.1%;等级≥3:1.5% vs 1.3%)。致命的出血事件、严重皮肤反应、血栓形成和穿孔发生频率低,双组之间的平衡,无论组织学。结论:我们证明添加N为非小细胞肺癌二线标准D治疗不会增加抗血管新生治疗相关AEs的频率,除了1 - 2级鳞状细胞癌患者的出血事件。这些AEs平衡无论组织学LUME-Lung 1。临床试验信息:NCT00805194。 UME-Lung 1,一项随机、安慰剂对照III期试验研究N + D在晚期非小细胞肺癌患者的一线化疗失败后,显著地提高PFS不论组织学,显示明显提高患者的腺癌生存。抗血管生成关联的 AEs的发病率和强度根据常见术语标准AEs(CTCAE版本3.0)评估所有病人至少接受一剂N,D,或安慰剂(Pl)。结果VEGF抑制剂AEs更常见(≥2%的差异)在N和Pl组出血(所有等级:14.1% vs 11.6%;等级≥3:2.3% vs 1.8%)和高血压(所有等级:3.5% vs 0.9%;等级≥3:0.6% vs 0.2%)。当我们比较组织学抗血管生成关联 AEs的差异时,我们发现了名义上的差异。更多流血事件被报道为N -治疗鳞状细胞癌(SCC)患者(所有等级:17.1% vs 10.9%;等级≥3:2.9% vs 1.3%)超过那些腺癌(所有等级:10.9% vs 11.1%;等级≥3:1.5% vs 1.3%)。致命的出血事件、严重皮肤反应、血栓形成和穿孔发生频率低,双组之间的平衡,无论组织学。结论:我们证明添加N为非小细胞肺癌二线标准D治疗不会增加抗血管新生治疗相关AEs的频率,除了1 - 2级鳞状细胞癌患者的出血事件。这些AEs平衡无论组织学LUME-Lung 1。临床试验信息:NCT00805194。
Lume-lung 2:多中心、随机、双盲、nintedanib III期研究,加上培美曲塞与安慰剂组加上培美曲塞非鳞非小细胞肺癌(NSCLC)后一线化疗的失败后晚期患者。(2013 ASCO)
摘要
背景:Nintedanib(N)是一种口服VEGFR,FGFR和PDGFR抑制剂。这个全球第三阶段的安全性和有效性研究调查了N +培美曲塞(PEM)与安慰剂(P)+ PEM的病人(pts)和晚期,非鳞 NSCLC之前曾接受化疗的患者。方法:随机分1:1 N 200毫克 口服 一天两次报+ PEM 500 mg / m2 静脉点滴 每21天(N = 353、组A)或P + PEM(N = 360、组B)。延续直到PD或N,P,PEM不可接受的毒性与,或二者联合是允许的。1°端点被集中审查的PFS。无效假设是在ITT人群测试发生后394事例(双面α=5%)。2°端点包括OS、研究者评价的 PFS,反应率(RR),安全,和生命质量。结果:基线pt特点是组 A和B之间是均衡的 (平均年龄59岁,女性45 - 42%,ECOG PS 1 62 - 61% , 腺癌95 - 93%, 贝伐单抗前8%)。根据研究者评价的PFS计划DMC无用分析,计划一千三,随机招募七百十三(确定没有安全问题)后停止。正在进行的患者进行揭盲和每个协议的后续随访。 1°端点(集中讨论PFS)随后的ITT分析的青睐A组向对B组(中位数4.4比3.6月,HR0.83[95%CI:0.7-0.99],P = 0.04)。N治疗过的患者疾病控制也显著的改善(61与53%,比值比为1.37,P = 0.039)。在OS(HR1.03)或RR(9%)无显着差异。探索性分析确定的时间从第一线疗法开始作为与N+ PEM(ASCO2013)改进的结果的预测指标的开始。N + PEM患者没有增加严重不良反应或不良事件。添加N+PEM导致≥G3 谷丙转氨酶ALT升高(23比7%),谷草转氨酶AST升高(12比2%),腹泻(3比1%)的发生率较高,但在≥G3高血压没有什么区别,出血,血栓形成,黏膜炎,或神经病变。结论:1即使研究提前终止。治疗N+ PEM与P+PEM相比较显著改善的先前接受化疗治疗晚期非鳞状非小细胞肺癌PFS,并有一个可管理的安全性。临床试验信息:NCT00806819。
Nintedanib(BIBF 1120)加上多烯紫杉醇在非小细胞肺癌患者一线化疗后进展: LUME Lung 1,随机,双盲III期试验。(2013 ASCO)
摘要
背景:Nintedanib(N)抑制VEGFRs PDGFRs,FGFRs。LUME Lung 1是局部晚期或转移性非小细胞肺癌一线治疗后进展N +多烯紫杉醇(D)的III期试验 (pts)控制的安慰剂。方法 IIIB:/ IV或复发性非小细胞肺癌(由组织学分层,ECOG PS,贝伐单抗之前,和脑转移)随机给予N 200毫克 每天两次+ D 75 mg / m2每21天(N = 655)或P + D(N = 659)。1°端点 713事例后集中讨论了PFS (log-rank双面分层l,α= 5%,β= 10%)。关键2°端点1121事例后分层次的分析了OS(2站,调整α= 4.98%,β= 20%),首先在腺癌(adeno)分< 9 mo一线疗法开始以来,(T < 9mo;认定为预后/预测生物标志物(ASCO 13]),然后所有腺的病人,然后所有的病人。预定义的敏感度分析靶病灶(SLD)的最长直径总和添加到分层因素的Cox模型。结果 t特点是组之间的平衡。N + DS与P + D相比较显著延长PFS(HR 0.79;CI:0.79,0.68,P = 0.0019;平均3.4 vs 2.7 mo)无论组织学(鳞状HR0.77,P = 0.02;腺 HR 0.77,P = 0.02)。OS显著延长所有腺癌 pts(HR 0.83;p = 0.0359;中位数12.6 vs 10.3 mo)中值最大的改进在T < 9mo 腺癌 pts(HR 0.75;p = 0.0073;中位数10.9 vs 7.9 mo)。有一种趋势,在所有患者以提高OS可见(HR =0.94,P=0.272,中位数10.1比9.1)。当调整SLD,一个显著OS获益见于所有患者(HR0.88,CI:0.78,0.99,P=0.0365)。N + D治疗所有的腺癌患者疾病控制率显著提高了(优势比[或]1.93;p < 0.0001),T < 9mo 腺癌患者(或2.90,p < 0.0001)和所有患者(或1.68,p < 0.0001)。最常见的副作用腹泻(任何:42.3 vs 21.8%;Gr≥3:6.6和2.6%)和谷丙转氨酶ALT升高(任何:28.5 vs 8.4%;Gr≥3:7.8和0.9%)。CTCAE的发生率 Gr≥3 AEs为71.3和64.3%。退出由于双组(21.7% vs 22.7)相似副作用,是Gr≥3高血压、出血或血栓形成。结论:N + D显著改善PFS,无论组织学,延长腺癌患者的OS。副作用可通过剂量减少和对症治疗控制。临床试验信息:NCT00805194。 t特点是组之间的平衡。N + DS与P + D相比较显著延长PFS(HR 0.79;CI:0.79,0.68,P = 0.0019;平均3.4 vs 2.7 mo)无论组织学(鳞状HR0.77,P = 0.02;腺 HR 0.77,P = 0.02)。OS显著延长所有腺癌 pts(HR 0.83;p = 0.0359;中位数12.6 vs 10.3 mo)中值最大的改进在T < 9mo 腺癌 pts(HR 0.75;p = 0.0073;中位数10.9 vs 7.9 mo)。有一种趋势,在所有患者以提高OS可见(HR =0.94,P=0.272,中位数10.1比9.1)。当调整SLD,一个显著OS获益见于所有患者(HR0.88,CI:0.78,0.99,P=0.0365)。N + D治疗所有的腺癌患者疾病控制率显著提高了(优势比[或]1.93;p < 0.0001),T < 9mo 腺癌患者(或2.90,p < 0.0001)和所有患者(或1.68,p < 0.0001)。最常见的副作用腹泻(任何:42.3 vs 21.8%;Gr≥3:6.6和2.6%)和谷丙转氨酶ALT升高(任何:28.5 vs 8.4%;Gr≥3:7.8和0.9%)。CTCAE的发生率 Gr≥3 AEs为71.3和64.3%。退出由于双组(21.7% vs 22.7)相似副作用,是Gr≥3高血压、出血或血栓形成。结论:N + D显著改善PFS,无论组织学,延长腺癌患者的OS。副作用可通过剂量减少和对症治疗控制。临床试验信息:NCT00805194。
|
 “砰”!国产新药获批,击穿肺癌靶向
作者:Tony
与欧美人种不同的是,亚洲肺癌患者EGFR突变率高,约达到50%[1],有更多机
“砰”!国产新药获批,击穿肺癌靶向
作者:Tony
与欧美人种不同的是,亚洲肺癌患者EGFR突变率高,约达到50%[1],有更多机
 父亲生病一周年小记
2024年3月9日父亲感觉吸气的时候胸部有压迫感去卫生院做了个CT,CT还要经上级医院审
父亲生病一周年小记
2024年3月9日父亲感觉吸气的时候胸部有压迫感去卫生院做了个CT,CT还要经上级医院审
 母亲奥西耐药后应如何选择
2022年10月确诊肺腺癌,基因检测结果EGFR21,服用奥希替尼。2024年6月CT影像检查缓慢
母亲奥西耐药后应如何选择
2022年10月确诊肺腺癌,基因检测结果EGFR21,服用奥希替尼。2024年6月CT影像检查缓慢
 又一个肺癌四代靶向药倒下,奥希替尼
作者:闵
自第一款EGFR-TKI易瑞沙(即吉非替尼)于2005年在我国上市以来,EGFR-TKI药
又一个肺癌四代靶向药倒下,奥希替尼
作者:闵
自第一款EGFR-TKI易瑞沙(即吉非替尼)于2005年在我国上市以来,EGFR-TKI药
 求助求助:妈妈的基因检测结果,EGFR
请版主帮忙@一下大神们,帮忙出一下注意。
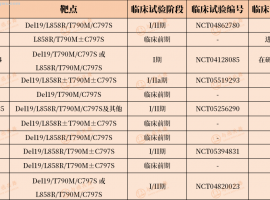
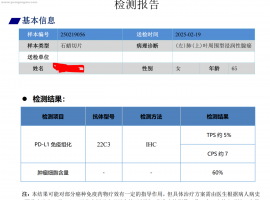
帖子更新到2025年3月7日,首次手术样本,送
求助求助:妈妈的基因检测结果,EGFR
请版主帮忙@一下大神们,帮忙出一下注意。
帖子更新到2025年3月7日,首次手术样本,送




 显身卡
显身卡



